Спинальная амиотрофия (спинальная мышечная атрофия или СМА) — это неизлечимое, почти всегда наследуемое заболевание, вызванное мутацией гена в 5-й хромосоме.
Мутация гена СМА приводит к недостатку белка, который необходим для построения белковых РНК-структур, и к недостаточному развитию поперечнополосатых мышц, преимущественно нижних конечностей, а также шейного отдела и головы.
Болезнь может проявиться с момента рождения, и даже когда плод еще в утробе, и в любой период жизни. У новорожденных спинальная амиотрофия чаще всего приводит к ранней смерти, но в некоторых случаях она может иметь мягкие формы, в основном в пожилом возрасте. Рассмотрим подробнее особенности этой патологии.
Спинальная амиотрофия — все о болезни
История и статистика
СМА — достаточно редкое заболевание, открытое немецким врачом Верднигом в 1891 г. Заболевает ею один человек из 6 — 10 тыс., однако носителем рецессивного гена СМА является каждый 50-й человек.
В 1898 г. Вердниг и еще один ученый Гофман установили, что причиной СМА является дегенеративное поражение и недостаточное количество двигательных (моторных) нейронов передних рогов спинного мозга — SMN (survival motor neurons).

Уже в 20-м веке (в 1956 г.) другие ученые Кугельберг и Веландер открыли менее злокачественную, с более мягкими проявлениями, форму СМА, которой болеют в ювенильном и взрослом возрасте.
Тип наследования при спинальной амиотрофии
Болезнь может наследоваться по любому из типов:
- аутосомно-доминантному;
- аутосомно-рецессивному;
- Х-сцепленному доминантному;
- Х-сцепленному рецессивному.
В связи с этим, классифицируется очень много разных форм СМА.
Детская форма СМА наследуется по аутосомно-рецессивному типу: если оба родителя — носители, то заболеет четвертая часть их потомства.
Аутосомно-доминантный тип наследования СМА приводит к проявлению болезни у детей с вероятностью 50%, даже если болен всего один родитель.
Виды спинальной амиотрофии
Спинальная мышечная атрофия разделяется на четыре формы:
- Младенческая (I) — спинальная мышечная атрофия Верднига-Гофмана: диагностируется с момента рождения до полугода.
- Промежуточная (II) — болезнь Дубовица: от семи месяцев до полутора лет.
- Юношеская (III) — б. Кюгельберга-Веландера: после полутора лет.
- Взрослая (IV): после 35 лет.
Симптомы спинально-мышечной атрофии
Общие симптомы при СМА:
- поражение проксимальных (средних) мышц и фасций;
- сохранение чувствительности в большинстве клинических случаев;
- задержки умственного и психического развития при спинальной мышечной амиотрофии крайне редки;
- возможна при некоторых видах атрофия не только мышц конечностей, но респираторных, жевательных и глотательных.

Степени тяжести СМА
- Самой тяжелой и неблагоприятной считается СМА первого вида (младенческая) — спинальная амиотрофия Верднига — Гоффмана, при которой дети не способны совершать активные движения, держать голову, самостоятельно сидеть. С большим трудом дается младенцу кормление, так как ему трудно сосать молоко и глотать его.
- Меньшей злокачественностью обладает болезнь Дубовица (II промежуточная форма СМА): при ней дети способны сидеть, держать голову и есть, однако ходить всё же неспособны.
- Наименее тяжелой является юношеская форма: несмотря на мышечную слабость, ребенок способен научиться ходить, однако заболевание хоть и медленно, но прогрессирует и может привести к ранней инвалидности.
- Четвертая взрослая форма СМА может привести из-за слабости м — ц проксимальных отделов к невозможности самостоятельного передвижения, выпадению рефлексов, но прогноз в отношение длительности жизни при этом остается благоприятным.

Другие виды спинально-мышечной атрофии
Кроме мутации в генах, вызывающих поражение проксимальных мышц, существуют подобные патологии, с разным типом наследования, приводящие к атрофии мышц и фасций дистальных (концевых отделов).
Список их довольно велик, сведем болезни в небольшую таблицу:
| Название СМА | Тип наследования | Особенности и симптомы |
| SMAX1 | Х -сцепленный рецессивный | Наблюдается в основном у пожилых, поражает бульбарные нервы черепа, вызывает нисходящий паралич. |
| SMAХ2 | Х — сцепл. рецессивный | Врожденная агрессивная форма, приводящая к смерти до 3 — х мес. Вызывает слабость, арефлексию, контрактуры и переломы. |
| SMAX3 | Х — сцепл. рецессивный | Поражает в основном мальчиков. Атрофия всех дистальных мышц. Медленное нарастание симптомов |
| Дистальная ДСМА1 | аутосомно — рецессивный | Врожденная, поражаются в основном руки, возможны тяжелые дыхательные нарушения |
| Дистальные формы ДСМА2 — ДСМА5 | аутосомно — рецессивный | Все четыре формы отличаются медленным прогрессированием, ДСМА5 диагностируется у молодых. |
| Дистальная СМА двух типов: VA и VB (DSMAVA и DSMAVB) | аутосомно-доминантный | Преимущественно атрофированы верхние конечности. |
| ДСМА тип 2D | аутосомно — рецессивный | Юношеское и взрослое заболевание с медленным развитием: поражаются и проксимальные, и дистальные м — цы вначале в голенях, затем в руках. |
| ДСМА тип 7А | аутосомно-доминантный | Очень редкая взрослая форма с поражением голосовых связок. |
| ДСМА тип 2А | аутосомно-доминантный | Разновидность болезни Шарко (аллельный тип) |
| Ювенильная SMA (тип HMN1) | аутосомно-доминантный | Встречается в юности |
| Врожденная спинальная амиотрофия | аутосомно-доминантный | Нарушение иннервации и атрофия м — ц бедер, стоп, коленей с контрактурой и деформацией; иногда поражается голосовые связки. |
| SMA Финкеля | аутосомно-доминантный | Начинается преимущественно в 35 — 37 лет, но зафиксированы случаи заболевания и в детском возрасте. Медленно развивается вначале в ногах, а затем в руках. Активность и рефлексы снижены, наблюдается непроизвольное дрожание (фасцикуляция). |
| SMA Джокела | аутосомно-доминантный | Поражаются у взрослых прокс. и дистальные м — цы. |
| SMA (тип LED1) | аутосомно-доминантный | Атрофия нижних конечностей у новорожденных. |
| SMA типа РМЕ | аутосомно — рецессивный | Атрофия дистальных мышц с нарушением иннервации и эпилептическими припадками |
| СМА с врожденными костными переломами | аутосомно — рецессивный | Тяжелые симптомы, как при болезни. Верднига-Гоффмана, отягощенные переломами. |
| СМА с гипоплазией | аутосомно-доминантный | Врожденная аномалия головного мозга с церебральными симптомами, микроцефалией и задержкой развития. |
| СМА ювенильная асимметричного типа | -------------- | Ею болеют молодые индийские мужчины |

В этой таблице следует обратить внимание на последние два вида спинальной амиотрофии:
- СМА с гипоплазией сопровождается отклонениями умственного и психического развития, что не характерно для остальных видом болезни.
- Асимметричная ювенильная (индийская) амиотрофия не передается по наследству. При этом заболевание после двух-пяти лет вялого течения может стабилизироваться. Симптомы фасцикуляции при этой специфической форме наблюдаются редко.
Лечение спинальной амиотрофии
Вылечить подобного рода болезни, как и любую наследственную патологию, затрагивающую спинной или головной мозг, кардинально невозможно. Эффективность многих используемых сегодня лекарств при терапии амиотрофии не доказана. Суть лечения сводится к увеличению белка, участвующего в формировании двигательных нейронов SMN.
Способы лечения СМА
Так, в основном применяются следующие препараты:
- вальпроевая кислота;
- оксибутират натрия;
- нусинерсен (новое лекарство, введенное в США в 2016 г. для лечения СМА).
Также необходимы поддерживающее лечение (ЛФК, массаж, физиотерапия), специальная белковая диета, при которой учитываются возможные противопоказания. Больные, лишенные возможности самостоятельного передвижения, нуждаются в социальной опеке.
Это наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1-1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии. Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани. Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
МКБ-10
G12.0 Детская спинальная мышечная атрофия, I тип [Верднига-Гоффмана]

Общие сведения
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Заболевание имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию - амиотрофию Кугельберга-Веландера . Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Причины
Амиотрофия Верднига-Гоффмана - наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) - ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы амиотрофии
Врожденная форма (СМА I) клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов . У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия , косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности , выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии , которая может явиться смертельно опасным осложнением спинальной амиотрофии .
Ранняя детская форма (СМА II) дебютирует после 6-месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
Амиотрофия Кугельберга-Веландера (СМА III) - наиболее доброкачественная спинальная амиотрофия детского возраста. Манифестирует после 2-х лет, в отдельных случаях в период от 15 до 30 лет. Отсутствует задержка психического развития , длительное время пациенты способны самостоятельно двигаться. Некоторые из них доживают до глубокой старости, не теряя способности к самообслуживанию.
Диагностика
В диагностическом плане для детского невролога имеет значение возраст появления первых симптомов и динамика их развития, данные неврологического статуса (в первую очередь наличие двигательных нарушений периферического типа на фоне абсолютно сохранной чувствительности), наличие сопутствующих врожденных аномалий и костных деформаций. Врожденная амиотрофия Верднига-Гоффмана может быть диагностирована неонатологом. Дифференциальный диагноз проводится с миопатиями , прогрессирующей мышечной дистрофией Дюшенна, боковым амиотрофическим склерозом , сирингомиелией, полиомиелитом, синдромом вялого ребенка , ДЦП , обменными заболеваниями.
С целью подтверждения диагноза проводится электронейромиография - исследование нервно-мышечного аппарата, благодаря которому выявляются характерные изменения, исключающие первично мышечный тип поражения и указывающие на патологию мотонейрона. Биохимический анализ крови не выявляет существенного повышения креатинфосфокиназы, характерного для прогрессирующей мышечной дистрофии. МРТ или КТ позвоночника в редких случаях визуализируют атрофические изменения передних рогов спинного мозга, но позволяют исключить другую спинальную патологию (гематомиелию , миелит , кисту и опухоль спинного мозга).
Окончательный диагноз «амиотрофия Верднига-Гоффмана» устанавливается после получения данных биопсии мышц и генетических исследований. Морфологическое изучение мышечного биоптата выявляет патогномоничную пучковую атрофию мышечных волокон с чередованием зон атрофии миофибрилл и неизмененной мышечной ткани, наличием отдельных гипертрофированных миофибрилл, участков соединительнотканных разрастаний. Проводимый генетиками ДНК-анализ включает прямую и косвенную диагностику. С помощью прямого метода возможна также диагностика гетерозиготного носительства генной аберрации, что имеет важное значение в генетическом консультировании сибсов (братьев и сестер) больных лиц, супружеских пар, планирующих беременность. При этом большую роль играет количественный анализ числа генов локуса СМА.
Дородовый анализ ДНК позволяет снизить вероятность рождения ребенка с болезнью Верднига-Гоффмана. Однако для получения ДНК-материала плода необходимо использовать инвазивные методы пренатальной диагностики: амниоцентез , биопсию хориона, кордоцентез . Амиотрофия Верднига-Гоффмана, диагностированная внутриутробно, выступает показанием к искусственному прерыванию беременности .
Лечение амиотрофии Верднига-Гоффмана
Этиопатогенетическая терапия не разработана. В настоящее время амиотрофия Верднига-Гоффмана лечится путем улучшения метаболизма периферической нервной системы и мышечной ткани с целью замедлить прогрессирование симптомов. В терапии применяют комбинации препаратов различных фармакологических групп: нейрометаболиты (препараты на основе гидролизата головного мозга свиньи, витамины гр. В, гамма-аминомасляная кислота, пирацетам), облегчающие нервно-мышечную передачу (галантамин, сангвинарин, неостигмин, ипидакрин), улучшающие трофику миофибрилл (глютаминовая к-та, коэнзим Q10, L-карнитин, метионин), улучшающие кровообращение (никотиновая к-та, скополамин). Рекомендована лечебная физкультура и детский массаж .
Современное развитие техники позволило несколько облегчить жизнь пациентов и их родственников, благодаря применению автоматизированных инвалидных колясок и портативных аппаратов ИВЛ. Улучшить подвижность пациентов помогают различные методы ортопедической коррекции. Однако основные перспективы в лечении СМА связаны с развитием генетики и поисками возможностей коррекции генетических аберраций методами генной инженерии.
Прогноз
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда - к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.
Частота заболевания
СМА – одно из самых часто встречеющихся заболеваний из орфанных (редких), болеет один новорожденный на 6000-10000 .
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.

От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III , болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА
IV
, этот тип называется ещё «взрослой СМА»,
поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
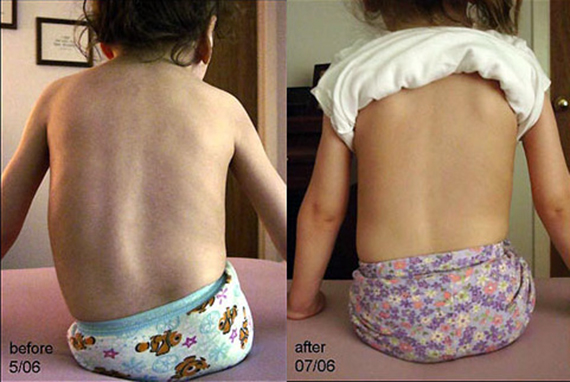
На сегодняшний момент радикального лечения от СМА не существует.
Международной корпорацией «Биоген» был разработан препарат «Спинраза», который значительно улучшил состояние больных, к которым применялся во время тестирования. В настоящее время препарат одобрен к применению в США, в Европе ориентировочная стоимость годового курса, по подсчётам компании, будет составлять около 270 тысяч евро, в России препарат не сертифицирован. Лечение пожизненное.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300 .
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?

Препарат «Спинраза», значительно улучшающий состояние больных. Фото с сайта healthbeat.spectrumhealth.org
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА

Итальянка Симона Спиноглио
родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен — о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов . Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий — член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.
Что такое Спинальная амиотрофия Верднига–Гоффманна
Спинальная амиотрофия Верднига-Гоффманна. Заболевание описано Дж. Верднигом в 1891 г. и Ж. Гоффманном в 1893 г. Частота 1 на 100 000 населения, 7 на 100 000 новорожденных.
Что провоцирует Спинальная амиотрофия Верднига–Гоффманна
Наследуется по аутосомно-рецессивному типу.
Патогенез (что происходит?) во время Спинальной амиотрофии Верднига–Гоффманна
Обнаруживаются недоразвитие клеток передних рогов спинного мозга, демиелинизация передних корешков. Часто имеются аналогичные изменения в двигательных ядрах и корешках V, VI, VII, IX, X, XI и XII черепных нервов. В скелетных мышцах нейрогенные изменения характеризуются «пучковой атрофией», чередованием атрофированных и сохранных пучков мышечных волокон, а также нарушениями, типичными для первичных миопатий (гиалиноз, гипертрофия отдельных мышечных волокон, гиперплазия соединительной ткани).
Симптомы Спинальной амиотрофии Верднига–Гоффманна
Выделяют три формы заболевания: врожденную, раннюю детскую и позднюю, различающиеся временем проявления первых клинических симптомов и темпом течения амиотрофического процесса.
При врожденной форме с первых дней жизни у детей выражены генерализованная мышечная гипотония и гипотрофия мышц, снижение либо отсутствие сухожильных рефлексов. Рано определяются бульбарные расстройства, проявляющиеся вялым сосанием, слабым криком, фибрилляциями языка, снижением глоточного рефлекса. Заболевание сочетается с костно-суставными деформациями: сколиозом, воронкообразной или «куриной» грудной клеткой, контрактурами суставов. Развитие статических и локомоторных функций резко замедлено. Лишь у ограниченного числа детей с большим опозданием формируется способность держать голову и самостоятельно садиться. Однако приобретенные двигательные навыки быстро регрессируют. У многих детей с врожденной формой болезни снижен интеллект. Часто наблюдаются врожденные пороки развития: врожденная гидроцефалия, крипторхизм, гемангиома, дисплазия тазобедренных суставов, косолапость и др.
Течение . Болезнь имеет быстро прогрессирующее течение. Летальный исход наступает до 9-летнего возраста. Одной из основных причин смерти являются тяжелые соматические расстройства (сердечно-сосудистая и дыхательная недостаточность), обусловленные слабостью мускулатуры грудной клетки и снижением участия ее в физиологии дыхания.
При ранней детской форме первые признаки болезни возникают, как правило, на втором по луго дии жизни. Моторное развитие в течение первых месяцев удовлетворительное. Дети своевременно начинают держать голову, сидеть, иногда стоять. Заболевание развивается подостро, нередко после инфекции, пищевой интоксикации. Вялые парезы первоначально локализуются в ногах, затем быстро распространяются на мышцы туловища и руки. Диффузные мышечные атрофии сочетаются с фасцикуляциями, фибрилляциями языка, мелким тремором пальцев, сухожильными контрактурами. Мышечный тонус, сухожильные и надкостничные рефлексы снижаются. В поздних стадиях возникают генерализованная мышечная гипотония, явления бульбарного паралича.
Течение . Злокачественное, хотя и мягче по сравнению с врожденной формой. Летальный исход наступает к 14-15 годам жизни.
При поздней форме первые признаки болезни возникают в 1,5- 2,5 года. К этому возрасту у детей полностью завершено формирование статических и локомоторных функций. Большинство детей самостоятельно ходят и бегают. Заболевание начинается незаметно. Движения становятся неловкими, неуверенными. Дети часто спотыкаются, падают. Изменяется походка: они ходят, сгибая ноги в коленях (походка «заводной куклы»). Вялые парезы первоначально локализуются в проксимальных группах мышц нижних конечностей, в дальнейшем сравнительно медленно переходят на проксимальные группы мышц верхних конечностей, мышцы туловища; атрофии мышц обычно малозаметны вследствие хорошо развитого подкожного жирового слоя. Типичны фасцикуляции, мелкий тремор пальцев, бульбарные симптомы - фибрилляции и атрофия языка, снижение глоточного и небного рефлексов. Сухожильные и надкостничные рефлексы угасают уже в ранних стадиях болезни. Костно-суставные деформации развиваются параллельно основному заболеванию. Наиболее выражена деформация грудной клетки.
Течение . Злокачественное, но мягче, чем у первых двух форм. Нарушение способности самостоятельной ходьбы происходит в 10-12-летнем возрасте. Больные живут до 20-30 лет.
Диагностика Спинальной амиотрофии Верднига–Гоффманна
Диагноз строится на основании данных генеалогического анализа (аутосомно-рецессивный тип наследования), особенностей клиники (раннее начало, наличие диффузных атрофии с преимущественной локализацией в проксимальных группах мышц, генерализованной мышечной гипотонии, фасцикуляций и фибрилляций языка, отсутствие псевдогипертрофий, прогредиентное и в большинстве случаев злокачественное течение и др.), результатах глобальной (накожной) и игольчатой электромиографии и морфологического исследования скелетных мышц, позволяющего выявить денервационный характер изменений.
Дифференцировать врожденную и раннюю формы следует в первую очередь от заболеваний, входящих в групп у синдромов с врожденной мышечной гипотонией (синдром «вялого ребенка»): амиатонии Оппенгейма, врожденной доброкачественной формы мышечной дистро фии, атоническо й формы детского церебрально го паралича, наследственных болезней обмена веществ, хромосомных синдромов и др. Позднюю форму следует дифференцировать от спинальной амиотрофии Кугельберга-Веландера, прогрессирующих мышечных дистрофий Дюшенна, Эрба-Рота и др.
Лечение Спинальной амиотрофии Верднига–Гоффманна
При спинальной амиотрофии Верднига-Гофмана назначают ЛФК, массаж, препараты, улучшающие трофику нервной ткани - церебролизин, аминалон (гаммалон), пиридитол (энцефабол).
К каким докторам следует обращаться если у Вас Спинальная амиотрофия Верднига–Гоффманна
Невролог
Акции и специальные предложения
Медицинские новости
14.11.2019
Специалисты сходятся во мнении, что необходимо привлечение внимания общественности к проблемам сердечно-сосудистых заболеваний. Некоторые из них являются редкими, прогрессирующими и трудно диагностируемыми. К таким относится, например, транстиретиновая амилоидная кардиомиопатия
14.10.2019
12, 13 и 14 октября, в России проходит масштабная социальная акция по бесплатной проверке свертываемости крови - «День МНО». Акция приурочена к Всемирному дню борьбы с тромбозами. 05.04.2019
Заболеваемость коклюшем в РФ за 2018 г. (в сравнении с 2017 г.) выросла почти в 2 раза 1, в том числе у детей в возрасте до 14 лет. Общее число зарегистрированных случаев коклюша за январь-декабрь выросло с 5 415 случаев в 2017 г. до 10 421 случаев за аналогичный период в 2018 г. Заболеваемость коклюшем неуклонно растет с 2008 года...
Медицинские статьи
Почти 5% всех злокачественных опухолей составляют саркомы. Они отличаются высокой агрессивностью, быстрым распространением гематогенным путем и склонностью к рецидивам после лечения. Некоторые саркомы развиваются годами, ничем себя не проявляя...
Вирусы не только витают в воздухе, но и могут попадать на поручни, сидения и другие поверхности, при этом сохраняя свою активность. Поэтому в поездках или общественных местах желательно не только исключить общение с окружающими людьми, но и избегать...
Вернуть хорошее зрение и навсегда распрощаться с очками и контактными линзами - мечта многих людей. Сейчас её можно сделать реальностью быстро и безопасно. Новые возможности лазерной коррекции зрения открывает полностью бесконтактная методика Фемто-ЛАСИК.
Косметические препараты, предназначенные ухаживать за нашей кожей и волосами, на самом деле могут оказаться не столь безопасными, как мы думаем
- Слабость в мышцах: неловкость при движениях, нарушение походки (шаг со сгибанием ног в коленях).
- Снижение тонуса мышц.
- Уменьшение конечностей в объеме из-за недоразвития мышц.
- Тремор (мелкое подергивание, тряска) конечностей.
- Снижение мимики.
- Нарушение глотания - поперхивание при глотании.
- Нарушение дыхания и, как следствие, частые бронхиты, пневмонии (воспаление легочной ткани). Возникает из-за нарушения работы дыхательной мускулатуры, при этом развивается застой крови в легких. Это является условием, предрасполагающим к развитию воспаления.
- Деформации костно-суставного аппарата: грудной клетки (« куриная», воронкообразная грудь), позвоночника (сколиоз – искривление позвоночника).
- Необратимое снижение подвижности в суставах конечностей (контрактуры).
- Замедление физического развития ребенка.
Формы
- Врожденная -
признаки наблюдаются сразу после рождения:
- ребенок вялый;
- плохо берет грудь;
- крик слабый;
- на окружение реагирует вяло;
- эта форма часто сочетается с признаками нарушения развития скелета (сколиоз - искривление позвоночника, деформация грудной клетки).
- Ранняя детская: ребенок до полугода развивается нормально, затем (часто после кишечной инфекции) развиваются параличи сначала ног, затем рук, тонус мышц снижается. Дети не держат головку, не сидят, не переворачиваются.
- Поздняя: первые признаки появляются к 1-3 годам, часто появление первых признаков остается незамеченным. Ребенок нормально развивается, затем начинает нарушаться походка, движения становятся неловкими, затем присоединяется общее снижение тонуса мышц.
Причины
- Заболевание является наследственным, то есть передается от родителей детям.
Диагностика
При появлении вышеописанных симптомов необходимо обратиться к или .
Врач выполнит полный осмотр ребенка, проведет неврологическое обследование, а также обсудит порядок и сроки развития признаков заболевания с родителями.
Важным является сбор данных о возможном наличии признаков заболевания у кого-то из семьи, что может натолкнуть врача на мысль о наследственной природе заболевания.
Для подтверждения диагноза врач назначает:
- ЭНМГ (электронейромиография): датчики на коже фиксируют прохождение нервного импульса. Метод позволяет определить, с чем связаны двигательные нарушения;
- биопсию мышц: для исследования иглой берется маленькая часть мышцы;
- МРТ (магнитно-резонансная томография) головы: для исключения другой патологии (метод позволяет послойно изучить строение любой части организма);
- генетический анализ (исследование строения хромосом, поиск генетических мутаций) - является методом, подтверждающим диагноз.
Лечение амиотрофии детской спинальной Верднига-Гоффмана
Методов или препаратов, воздействующих на причину заболевания, на данный момент нет.
Применяются методы, способствующие улучшению питания нервной системы:
- витаминотерапия;
- ноотропы (препараты, улучшающие питание мозга);
- массаж;
- лечебная физкультура.
Осложнения и последствия
- Обездвиженность.
- Остановка дыхания.
- Риск летального исхода.
Течение ранней формы более мягкое: смерть наступает в 14-15 лет.
При поздней форме дети теряют способность ходить к 10-12 годам, живут до 20-30 лет.
Профилактика амиотрофии детской спинальной Верднига-Гоффмана
- Ввиду наследственного характера заболевания профилактика невозможна.